
Biodistribution, COVID mRNA Gene Therapy/Vax
Vax makers didn't commit fraud, they delivered the fraud that the USG ordered
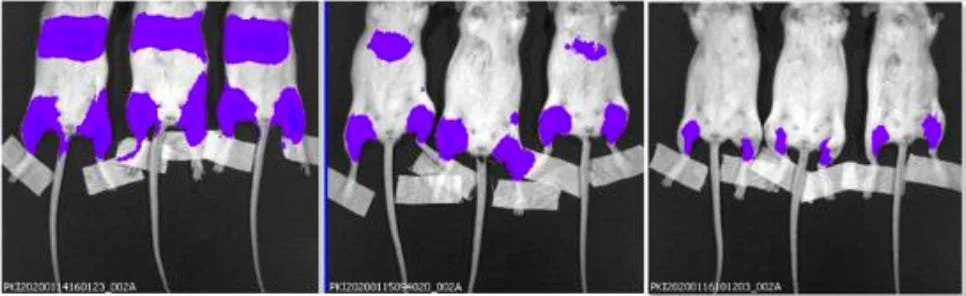
Figure 2: Bioluminescence measurement in the liver using the LNP-formulated modRNA encoding for luciferase
BALB/c mice were injected i.m. in the right and left hind leg with each 1 μg of LNP-formulated modRNA encoding luciferase. A) At 6 h, 24 h, and 48 h after injection, the luciferase expression in vivo was measured by luciferin application

Bioluminescence Measurements
The biodistribution of luciferase expressed by the LNP-formulated modRNA after i.m. injection was assessed by bioluminescence measurements. Mice received a total dose of 2 μg LNP8.
Usual Disclaimer: The opinions in this essay are those of the authors, and do not necessarily represent those of the United States Government (USG), Centers for Disease Control and Prevention (CDC), or the Advisory Committee on Immunization Practices (ACIP).
This ACIP exchange with Pfizer and Moderna captured in the clip above is the topic for today’s Substack essay.
The key issue here is the promoted lie that the COVID mRNA products stay at the site of injection and do not distribute throughout the body of those who accept these injections. We now have documentation that this was known to be false information even before the clinical trials were initiated because of studies performed and results reported to FDA and other global regulatory authorities in the regulatory documents (IND or CTD) filed by Pfizer and Moderna as a requirement before initiating human clinical trials. The technical terms used to describe this general area of pharmaceutical and regulatory science are “pharmacodistribution” (where the drug goes) and “pharmacokinetics” (how long the drug sticks around). Industry slang for this is PD and PK studies. Pharmacodistribution is also known as Biodistribution (BD).
What was revealed at the recent ACIP meeting was that neither Moderna nor Pfizer/Bio-N-Tech performed PD or BD studies for the COVID mRNA products in compliance with internationally established standards, and in the case of Pfizer, the Pfizer ACIP representative indicated that the manipulation and editing of the PD/BD data submitted to the FDA was done in coordination and with the full awareness of the FDA. Speaking as a regulatory specialist with experience in developing, submitting, and negotiating IND applications to the FDA, I personally find this admission stunning. There is supposed to be a firewall between the sponsor (Moderna or Pfizer) and the regulators. Not a cooperative teaming relationship.
FDA nonclinical biodistribution guidance for industry for Gene Therapy Products can be found at this link.
DEFINITION OF NONCLINICAL BD (Biodistribution)
BD is the in vivo distribution, persistence, and clearance of a GT product at the site of administration and in target and nontarget tissues, including biofluids (e.g., blood, cerebrospinal fluid, vitreous fluid). Nonclinical BD assessment entails the use of analytical methods to detect the GT product and transferred genetic material in collected samples and can include methods to detect the expression product of the transferred genetic material.
TIMING OF NONCLINICAL BD ASSESSMENT
BD data should be available when evaluating and interpreting the nonclinical pharmacology and toxicology findings. Nonclinical BD data can also inform design aspects of a first-inhuman clinical trial. It is important that nonclinical BD assessments be completed prior to initiation of the clinical trial.
Test Article
The test article administered in the nonclinical BD studies should be representative of the intended clinical GT product, taking into consideration the manufacturing process, important product characteristics (e.g., titer), and the final clinical formulation. In some situations, nonclinical BD data generated with a GT product consisting of the same vector intended for clinical use and a different therapeutic transgene or an expression marker gene (e.g., adeno-associated virus vector of the same serotype and promoter that directs expression of a fluorescent marker protein transgene) can be leveraged to support the BD profile.

Pfizer (and thus the FDA; many of the documents say “FDA: CONFIDENTIAL” at the lower boundary) knew that, contrary to what the highly paid spokesmodels and bought-off physicians were assuring people, the mRNA, spike protein and lipid nanoparticles did not stay in the injection site in the deltoid, but rather went, within 48 hours, into the bloodstream, from there to lodge in the liver, spleen, adrenals, lymph nodes, and, if you are a woman, in the ovaries.
“Pfizer vaccine fraud: Government was involved in dismissing accusations”
Regarding the statement from Moderna during the recent ACIP meeting that their animal (non-clinical) biodistribution study data were obtained using the same drug substance used in the subsequent vaccine product for human use, reporter Maryanne Demasi, PhD has documented that the statement of the Moderna representative to the ACIP was not factual.
Will there be any consequences for a vaccine manufacturer if it lies under questioning by the ACIP?
As summarized by Demasi, “When ACIP finally confronted Moderna and Pfizer, both companies evaded questions — and Moderna lied outright.”

There are several issues related to the irregular aspects of the PK and PD/BD data submissions provided to the FDA by Moderna and Pfizer. It has taken me four years to get to the point where I could directly question these companies about these irregularities, but I finally got the chance at the most recent ACIP meeting.
I became aware of these issues and irregularities in 2021 consequent to reviewing the Japanese “Common Technical Document” (equivalent to the FDA Initial New Drug application submitted by Pfizer) that had been identified and downloaded by Canadian Vaccinologist Dr. Byram Bridle, as well as review of the the European Medicines Agency (EMA) Assessment Report on Comirnaty (COVID-19 mRNA vaccine) issued on 19 February 2021. The EMA document review was performed at the request of Trial Site News, with key observations and findings published May. 28, 2021 in an article titled “Did Pfizer Fail to Perform industry Standard Animal Testing Prior to Initiation of mRNA Clinical Trials?”
To obtain independent reviews of these EMA regulatory documents, TrialSite contacted both Dr. Robert W. Malone, MD, MS, and another expert that wished to remain anonymous, and provided them copies of the EMA analysis and the FOIA documents. Dr. Malone was the original inventor of the mRNA vaccine technology back in the late 1980s. He currently advises several companies in regulatory affairs and clinical development. One of TrialSite’s other sources is a senior regulatory specialist who currently serves as the President of a prestigious European association. When asked to review and comment on the EMA assessment, Dr. Malone noted that normal pharmacokinetic and pharmaco-toxicology studies had not been performed before EUA authorization for the product. “I was particularly surprised that the dossier of regulatory documents indicates allowance for use in humans based on non-GLP PK and Tox studies relying on formulations which are significantly different from the final vaccine.“ After completing a review, TrialSite’s other source noted the following:
“A quick review the Toxicology Section (2.3.3) of The European Medicines Agency (EMA) Assessment Report on Comirnaty (COVID-19 mRNA vaccine) issued on 19 February 2021, raises concerns about data applicability of preclinical study findings to clinical use:
To determine the biodistribution of the LNP-formulated modified mRNA (modRNA), the applicant did study distribution of the modRNA in two different non-GLP studies, in mice and rats, and determined the biodistribution of a surrogate luciferase modRNA.
Thus, one might question the validity and applicability of non-GLP studies conducted using a variant of the subject mRNA vaccine.
In addition, no genotoxicity data were provided to EMA.”
Based on the FOIA documents, the biodistribution results (which are not disclosed in the public EMA summary document) suggest that the delivery technology results in mRNA delivery and significant concentration of the delivery lipids in ovaries, spleen, and other tissues and organs.
It is useful to keep in mind that, prior to these submissions, the vaccines group at FDA/CBER had very little experience and expertise relating to this (mRNA-based gene therapy) technology. There was relevant expertise in the FDA/CBER gene therapy group, but they seem to have been siloed off from providing regulatory advice and input to those overseeing the COVID mRNA vaccine submissions. This practice was highly unusual in my experience; the norm has always been that branches of the FDA with relevant experience and expertise are always brought in to complement the group with primary review responsibilities.
This also relates to the aggressively promoted and counterfactual denial that the technology and formulations used for the COVID mRNA “vaccine” products were actually gene therapy technology applied for a vaccine indication. If Dr. David Marks (FDA CBER Director) and his colleagues had acknowledged the obvious truth, then established regulatory guidance for gene therapy products would have been applicable to the COVID mRNA “vaccine” products. If classified as gene therapy technology applied to vaccination, this would have required more rigorous (and time-consuming) testing of risks, including genotoxicity and shedding.
Regulatory standards for products classified as “vaccine products” are much more lax than for those classified as ‘gene therapy products”. That is a more general problem that needs to be addressed as soon as possible. Why should “vaccine products” be treated less rigorously than other biologicals, when unlike others they are typically administered to patients that do not have a disease?
Basically, Dr. Peter Marks and colleagues used a contrived argument to avoid and bypass FDA policies that would have been problematic for the agenda they shared with the sponsors (Pfizer/Bio-n-Tech and Moderna) to jam these products through the regulatory review process as fast as possible by bypassing internationally established relevant regulatory standards that had been developed over decades.
After reviewing and analyzing the Japanese submission and EMA data on behalf of Trial Site News, I was struck by the many regulatory irregularities identified in the document, including those relating to the PK and PD studies. The sponsors were not following the established international regulatory standards. My working assumption was that the vaccine developers/manufacturers (sponsors) were misleading naive regulatory authority reviewers, and that those reviewers lacked the necessary background and experience to thoroughly review the technical details of the studies and data being presented.
So, based on that assumption, I wrote to Dr. Peter Marks (then Director, FDA/CBER) and offered my assistance and expertise as the person who had developed much of this tech - including the use of the luciferase reporter gene/protein in animals for identifying quantity and distribution of foreign gene delivery product.
Here is the text of the June 03, 2021 email setting up the meeting. I include it below just to provide context and demonstrate how I (naively) approached the FDA over these issues.
Hi Peter
Thanks for your reply.
(good to meet you electronically, Lorrie)
Unfortunately, the ACTIV meeting is at that time, and I really want to learn what has happened with the IRB dust up for the ACTIV-6/ivermectin arm!
That is looking like a bit of a mess. Fortunately for you, it is a CDER issue...
Attached is my calendar for today. Can we find time to fit anything into that?
I understand that Janet <Woodcock> has been talking to Steve <Kirsch> about these same general issues.
But he has been getting a bit wound up, is way outside of his core competencies, and lacks perspective.
I also understand that others within the agency share at least some of my concerns and that there are internal discussions ongoing.
Please understand that I do have some deep insight into the underlying technology (CV attached). This may or may not be duplicated in your division.
At this point, I am merely expressing concern based on the limited data which has been made available to me.
I hope and trust that the agency has more extensive information, and that it has required Pfizer/Biontech to continue to characterize the pharm/tox (beyond what is documented in the Japanese IND and the EMA assessment) during this period when the product is still under review despite the EUA.
Meaning that I trust that you have more data than I have been able to access and review.
I have faith that your division will do its work - it is not my objective to second guess.
And my friends within the agency speak highly of you - that is sincere truth.
for what it’s worth.
Very best wishes, with gratitude, and looking forward to your reply.
robert
I was so naive and trusting back then, defaulting to an assumption of professionalism and good intent. Trying to show respect and a willingness to be helpful rather than confrontational and accusatory. Trying to be a “nice guy.” Silly me.
During the following meeting, Dr. Marks listened to my concerns and related that he had seen additional data from Pfizer (those would be the documents finally released on court order, often referred to as “The Pfizer Papers”). Dr. Marks informed me that he had reviewed the documents, found nothing concerning, and requested that I extend professional courtesy by holding off on further publicizing my concerns until the FDA had an opportunity to address them in public statements.
Once the documents were made available (which required a lawsuit and court order), the analysis resulted in 50 reports, collectively known as the “Pfizer Files,” published in book form as Pfizer Documents Analysis Reports starting in January 2022, with wider public release in early 2023.
Key claims from the analysis include that Pfizer allegedly knew the mRNA vaccines were ineffective, with reported efficacy based on only 170 COVID-19 cases out of over 40,000 participants, leading to a real-world effectiveness estimate of less than 50%. The reports also claim that vaccine components, including lipid nanoparticles (LNP), distribute throughout the body and accumulate in organs such as the liver, adrenal glands, spleen, and ovaries.
Other allegations include that Pfizer and the FDA were aware of potential heart damage in minors, yet delayed public warnings; that the company hired thousands of additional staff to manage over 158,000 adverse event reports within the first 12 weeks of rollout; and that there were high rates of fetal death—87.5% in reviewed cases—among pregnant women who reported adverse events.
The documents reportedly indicate that Pfizer acknowledged the possibility of “shedding,” where unvaccinated individuals could be exposed to vaccine components via skin contact, inhalation, or sexual transmission, contradicting CDC statements dismissing shedding as a myth. Additionally, concerns were raised about impacts on fertility, with evidence suggesting accumulation of nanoparticles in reproductive organs and lack of proper toxicity testing in this area.
This history clearly documents active collusion between Dr. Peter Marks, representing the US Government Food and Drug Administration Center for Biologics Evaluation and Research and Pfizer to mislead the US public concerning the safety, efficacy, and Pharmacodistribution (PD)/Biodistribution (BD) of the mRNA vaccine products. From a legal perspective, this completely negates any possibility of a lawsuit or whistleblower action on behalf of the US Government against Pfizer (or Moderna) for their actions in this matter, because it is clear that the FDA colluded with the manufacturers - as Pfizer specifically admitted during the recent ACIP meeting.
This is the significance of the responses provided to the ACIP upon recent questioning about whether biodistribution data had been manipulated and/or were not performed to globally accepted regulatory standards (such as using the actual active drug substance rather than a surrogate, and meeting established GLP/GMP performance standards).
Moderna lied to the ACIP, and Pfizer essentially stated that their manipulations and misrepresentations were done with the full knowledge of the FDA.
Quoting Dr. Maryanne Demasi’s research -
https://www.fda.gov/media/155931/download?attachment
The Food and Drug Administration’s (FDA) own approval summary is clear:
“A biodistribution study was not performed with mRNA-1273 vaccine. Results from the biodistribution study of a different vaccine… were submitted in support of Spikevax.”
In response to direct ACIP questioning, Pfizer admitted to colluding with the FDA to manipulate data, and Moderna lied.

Where did this Biodistribution question to Pfizer come from?
Canadian vaccinologist Dr. Byram Bridle had recently completed a rigorous retrospective assessment of the initial Pfizer submission to the Japanese regulatory authority, with a particular focus on the Pharmacodistribution/Biodistribution submission information. He then shared his findings with me. The following is a summary of his sworn statements concerning his findings, which span two different reports.
Report #1- Report on Pfizer Biodistribution Studies to Highlight Four Instances of Potential Fraud.
Background
1. One type of experiment that is performed with vaccines to assess their safety is what is known as a biodistribution study. The purpose is to ascertain the anatomical distribution and kinetics of removal from the body of a novel medical intervention; in the case of modified RNA vaccines (modRNA), this would be cationic lipid nanoparticle-encapsulated modRNA.
2. A request was made for the United States Food and Drug Administration (FDA) to publicly disclose the data package that formed the basis for them to issue Emergency Use Authorization for the rollout of COVID-19 vaccines. The FDA requested permission to release these data over a period that would extend between 55 and 75 years. However, a court-issued order required the FDA to expedite the release of the documents. What ensued was a series of ‘data dumps’, in which large amounts of documentation were sequentially released to the public.
3. Within the first data package were two common technical documents that Pfizer/BioNTech had submitted for its COVID-19 vaccine (BNT162, also known as Comirnaty). They described preclinical biodistribution studies. One document described the results of an experiment conducted with mice. The other document reported results with rats. Both studies assessed cationic lipid nanoparticle-encapsulated modRNA that was intended to be analogous to the COVID-19 vaccine destined for use in people. The murine study that was released by the FDA was extensively redacted.
4. Prior to the FDA releasing these two common technical documents describing the biodistribution studies, a single document that provided descriptions of both studies had been submitted by Pfizer/BioNTech to Japan’s Pharmaceuticals and Medical Devices Agency. The Japanese government had made this document publicly available on their website.
Purpose
5. In this report, Dr. Bridle uses a head-to-head comparison of data provided in the different common technical documents describing the same biodistribution studies to highlight four instances of potential fraud.
Preamble
12. Websites that are referenced in this report were confirmed to be accurate as of August 12, 2025.
13. The biodistribution studies that are appended to this report raise numerous scientific concerns that were not properly addressed prior to the public rollout of modRNA COVID-19 vaccines. This includes the fact that one of the methods used to generate some of the data (i.e., bioluminescence imaging) is one of the most insensitive techniques for assessing biodistribution. This means that the studies were designed with an inherent bias towards minimizing the detection of systemic biodistribution of the novel modRNA products.
However, this report will not focus on these kinds of scientific nuances. Instead, this report focuses on four examples of potential fraudulent reporting that were identified by the author.
Potentially Fraudulent Reporting of modRNA Vaccine Biodistribution Data
Timeline
14. Dr. Peter Doshi from the University of Maryland, USA, published a paper in the scientific journal The BMJ on May 18, 20213. Reference #25 in the article pointed to the Japanese common regulatory document describing two of Pfizer/BioNTech’s preclinical studies in which the biodistribution of a surrogate modRNA vaccine was evaluated in mice and rats.
This report had been submitted to Japan’s Pharmaceuticals and Medical Devices Agency and is attached hereto and marked as Appendix “B”. The publication in The BMJ is the first known public reference to the Japanese version of the biodistribution study.
15. The FDA released its versions of Pfizer/BioNTech’s two biodistribution studies (one in mice, one in rats) as two separate common technical documents in the first week of March 2022.
16. The common technical document released by the FDA that describes the biodistribution study that used mice is attached hereto and marked as Appendix “C”.
17. The common technical document released by the FDA that describes the biodistribution study that used rats is attached hereto and marked as Appendix “D”.
18. Pfizer/BioNTech’s report on their biodistribution study was removed from the website of Japan’s Pharmaceuticals and Medical Devices Agency shortly thereafter; sometime between March 26, 2022, and April 14, 2022.
Potential Fraud #1 (Manipulation of data and lying about results)
19. A surrogate modRNA vaccine was tested in mice. It incorporated a modRNA encoding a gene for a protein known as luciferase. Following injection, mice could be treated such that anywhere the modRNA went, the luciferase would emit a light signal that could be detected (with low sensitivity) using specialized external whole-body imaging of the live rodents under anesthesia.
20. Page 19 (Section 5.1 Bioluminescence Measurements) of the murine study released by the FDA (Appendix “C”) shows the following highly redacted information:

21. Dr. Bridle recognized that the single image shown in the FDA’s document was a portion of an image from page 7 of the Japanese common technical document (Appendix “B”), as shown here:
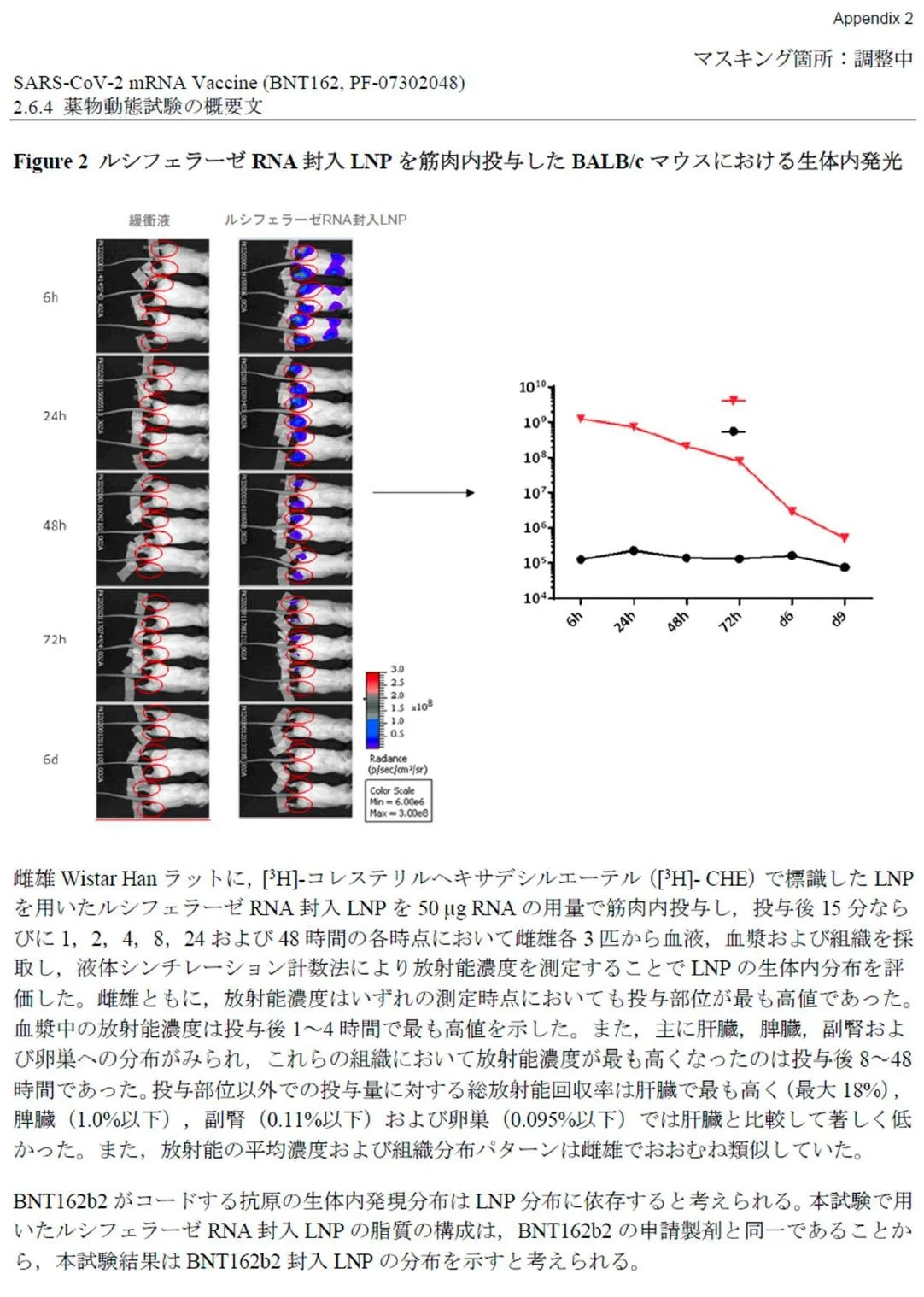
Specifically, the image in the FDA’s document is a portion of the upper image in the second column of the Japanese document (six-hour timepoint).
A close-up comparison of these two images revealed multiple concerns. The following annotated comparison of the images was produced by Dr. Bridle

23. Note that in the images shown above, the mice are lying on their bellies, so it is their backs that are showing.
24. A typical luminescence scale was added to the left side of the image, and an illustration of the anatomy of a mouse was added to the right side.
25. The image from the FDA’s version of the document is on the left and beside it is the image from the Japanese document from which the former appears to have been derived.
26. In this experiment, the site of injection of the modRNA vaccine was the large muscles of the upper hind limbs of the mice (not the shoulder muscle, as is commonly used in people).
27. Dotted red lines were added to the images to indicate where the cropping appears to have occurred.
28. An analysis of the two images seems to indicate that they represent identical mice, based on characteristics like the shape of the luminescent signals, the positions of the tails of the mice, the positions of their feet and the tape applied to the lower hind limbs, etc.
29. Several manipulations are evident. First, the image from the FDA was isolated from any relevant context. Specifically, in the Japanese document the image appeared alongside images from control mice, and from various timepoints.
30. Second, the image was rotated 90o relative to how it appeared in the Japanese document.
31. Third, the intensity of the luminescence signal was dialed down. This can be done with any bioluminescent image using the software used for acquisition of the image. When such a manipulation is done, one would expect a different file name to be applied, which is evident by the different labeling in the bottom left corners of the images.
32. Finally, and of substantial concern, the image has been cropped, apparently to exclude visualization of an obvious luminescent signal in the region of the kidneys and adrenal glands.
33. In summary, the image in the FDA’s version of the common technical document appears to be a version of the image from the Japanese document that was manipulated in numerous ways, potentially to discourage discovery that they are one and the same. It was then cropped to hide clear evidence of systemic biodistribution of the modRNA vaccine.
34. It is important to note that the images in the Japanese common technical document were also cropped to exclude approximately one-quarter of the upper bodies of the mice, including their heads. This is atypical since the convention for publishing this kind of scientific data, where systemic distribution is being evaluated, is to show the entire body of the animal. It is particularly important to know whether a novel medical intervention distributes into the brain, especially for something that is not intended to go to that location.
One cannot rule out distribution to anatomical locations for which no data are shown.
35. The regulatory scientist(s) that reviewed these data should have questioned images that were cropped in this fashion and should have requested disclosure of uncropped images.
Page 21 (Section 5.2) of the FDA’s version of the murine biodistribution study (Appendix “C”) shows the following

36. In this case, the images show mice lying on their backs, so it is their bellies that are showing. This reveals a luminescent signal in the region of the liver. This indicates that the modRNA vaccine got distributed from the injection sites in the hind limbs to the liver.
37. Page 6 (Section 1 Summary) of the FDA’s version of the murine biodistribution study (Appendix “C”) includes the following text (yellow highlighting added for emphasis). There was no mention of the drainage to the region of the kidneys and adrenal glands, which seems to have been facilitated by cropping out the signal in the images that showed this.

38. It is important to note that this stated conclusion in the FDA’s document matches the public-facing messaging that claimed modRNA vaccines stayed at the injection site but were ultimately eliminated from the body via the liver. The liver could not be ignored as a site to which the modRNA vaccines distributed because regulatory documents had consistent tabulated data showing that the liver was the site with the highest concentration of the modRNA vaccines beyond the injection site.
39.Ultimately, the conclusions drawn about the biodistribution data in the FDA’s version of the mouse-focused common technical document contradicts the obvious conclusions that must be drawn unredacted and lesser-cropped images from the Japanese version.
Potential Fraud #2 (Using redaction to support a lie about clearance from the body)
40. Another important purpose of the murine biodistribution study was to ascertain how long the novel modRNA vaccine remained in the body. It is important from a regulatory perspective to show that a medical intervention that is intended to be present only transiently in the body, is ultimately eliminated.
41. Page 20 of the FDA’s version of the murine biodistribution study (Appendix “C”) includesnthe following figure panels and figure legend (yellow highlighting added for emphasis):

42. Note the claim that the luminescence signal had reached background levels by nine days following injection of the surrogate modRNA vaccine. Panel A of this figure was already shown in this report, above, when discussing Page 19 (Section 5.1 Bioluminescence Measurements). It did not show any data beyond six hours post-injection. That lack of transparency did not allow this conclusion to be evaluated. Further, panel B has been redacted so only the luminescent signal from control (sham-treated) mice can be evaluated. The curve showing the bioluminescent signal in mice that received the surrogate modRNA vaccine was completely hidden. Again, this did not allow an assessment of the claim the signal had reached background levels by day 9.
43. The claim that the modRNA was eliminated from the body within nine days of injections aligned with public messaging that this technology was short-lived in the body.
44. Dr. Bridle remembered seeing this same graph in the Japanese version of the common technical document (page 7 of Appendix “B”), which is shown here (on the right) alongside the redacted version from the FDA’s document (on the left), with annotations added for clarity:

45. It is clear from this comparison that the luciferase signal had not reached baseline by day nine post-immunization. An absence of error bars suggests that the number of experimental and/or biological replicates were insufficient to conduct a proper statistical analysis of the data. Such a truncated, data-deprived figure is suggestive of an overly rushed or preliminary experiment that should not have been used to support any firm conclusions.
46. The regulatory scientist(s) that reviewed these data should have requested that the study be repeated with an extended timeline and with sufficient experimental replicates until proper statistical analyses revealed a timepoint at which the luciferase signal in the immunized group was no longer statistically different from the background signal in the sham-treated control group. Apparently, this was not done.
47. Based on the data from the Japanese document, the claim in the FDA’s report that “After 9 d, the reporter expression dropped to background levels” appears to be a bald-faced lie that was facilitated by redacting data to avoid scrutiny and then hoping nobody would realize that non-redacted data had previously been released by Japan’s Pharmaceuticals and Medical Devices Agency.
Potential Fraud #3 (Failing to disclose the results of an initial study that raised serious concerns)
48. A comparison of the FDA’s common technical document describing the biodistribution study performed in rats (Appendix “D”) with the version submitted to Japan’s Pharmaceuticals and Medical Devices Agency (Appendix “B”) appears to reveal a failure to disclose scientific data to the Japanese government that would be pertinent to regulatory review.
49. The FDA’s version of the biodistribution study contains much more information than the Japanese version. The Japanese document implied that a single dose of the surrogate modRNA vaccine had been evaluated, with 50 μg of modRNA administered intramuscularly to each rat.
50. However, the FDA’s version (Appendix “D”) showed that there were two iterations of the experiment. An initial experiment had been attempted in which only male rats received 100 μg of modRNA. This initial study was never disclosed in Japan’s document (Appendix “B”).
51. Importantly, the initial study was a failure due to overt toxicity. The experiment was truncated because the 100 μg dose caused obvious harm to the rats. Section 7.1 on page 19 of the FDA’s document (Appendix “D”) draws the following conclusions

52. On page 11 of the same document (Appendix “D”), the study director noted that “a subsequent review of the data showed concentrations [of LNPs] were well detected in tissues”.
53. The treatment caused an acute body weight loss of ~7%.
54. Of the three males that were due to be euthanized at 48-hours post-inoculation, the treatment proved lethal for one (33.3% lethality), which was euthanized at 30 hours. Of the remaining two rats, they were showing clear signs of distress at 48 hours. As stated, “Additionally, animals 019M and 020M were hunched and piloerect from approximately 30 h post-dose onwards”. “Onwards” means up to 48 hours when they were euthanized to harvest tissues. In other words, 3/3 (100%) of the rats were clearly sick by 30-hours postimmunization and there is no way of knowing whether this would have got worse or resolved in the remaining two rats beyond 48 hours.
55. Only males were tested at the 100 μg dose, so it is unknown what it would have done to females. This is particularly important because it is noted that at the 50 μg dose in the second study, one of the females at the last (48-hour) timepoint was obviously sick, including signs of respiratory distress.
56. The study director contacted the study sponsor to tell them the experiment was failing and the decision rendered was to repeat it at half the dose. No consideration was apparently given to testing multiple doses, nor was expansion of the scope of the study done, despite obvious safety concerns, and signals that the experiment was too short.
57. The regulatory scientist(s) that reviewed these data should have been concerned about the serious safety signals in the initial study and should have requested expanded experimentation, including testing more doses for a longer duration.
58. Remarkably, it appears that there was a failure to disclose these pertinent facts to Japan’s Pharmaceuticals and Medical Devices Agency (their document makes no reference to this information) and disclosure by the FDA only came via court order.
Potential Fraud #4 (Hiding a serious and unique concern for females)
59. The FDA’s common technical document describing the biodistribution study performed in rats (Appendix “D”) revealed a second major data set that appears to have been withheld from the version submitted to Japan’s Pharmaceuticals and Medical Devices Agency (Appendix “B”).
60. Specifically, the Japanese document acknowledged that male and female rats were included in the biodistribution study in which a dose of 50 μg of modRNA was administered (now known to be the second of two studies to replace the initial failed experiment).
However, data showing accumulation of the modRNA vaccine in various tissues were not separated by sex in the Japanese document, except for analyses of ovaries and the uterus, which are obviously exclusive to females. Instead, for all other tissues (except the male specific prostate gland and testes), data from males and females were pooled.
61. This pooling of data is notable because the purpose of the study was to identify when peak concentrations of the cationic lipid nanoparticles from the modRNA vaccine could be detected in tissues. This would inform where the vaccine was going in the body and how long it remained there.
62. However, the study failed to identify peaks, which should have prompted regulatory scientists to request longer timepoints. In a state of crisis, one might contend that capturing a plateau may be sufficient. Then one could argue that there was at least no evidence of ongoing bioaccumulation. Indeed, data in the Japanese document (Appendix “B”) appeared to have captured plateaus in bioaccumulation for at least some tissues. For example, Dr. Bridle graphed the bioaccumulation data for the liver using the data from the Japanese document, as shown here

The red line shows the graph has approximated a plateau.
63. However, the data from the FDA’s document, which separated the data by sex, showed the following sex-specific kinetics:

64. The lipid nanoparticles from the modRNA vaccine accumulated rapidly in the males and was declining by the end of the experiment (48 hours post-immunization). In contrast, the concentration was spiking in the liver of females at the last timepoint.
65. Consequently, Dr. Bridle graphed the kinetics of bioaccumulation of the lipid nanoparticles from the modRNA vaccine in females based on the data from the FDA’s document. These were the results from fourteen other tissues


Red arrows consistently show concentrations of lipid nanoparticles spiking, in some cases exponentially or near-exponentially in all fourteen tissues.
66. These are disconcerting results that suggest there is a unique eect of female biology on the biodistribution of modRNA vaccines.
67. This should have served as a major safety signal to trigger more extensive research to understand this unique outcome in females.
68. An obvious question to pose would be, ‘how high would the concentrations go and how long would these lipid nanoparticles remain in the body of a female?’.
69. No regulatory scientist should have approved the use of this technology in women and/or girls, let alone allowing them to be mandated in these demographics with these kinds of data in-hand.
70. Remarkably, it appears that this information was not disclosed to Japan’s Pharmaceuticals and Medical Devices Agency (it seems to absent from Appendix “B”). And, again, disclosure by the FDA only came via court order.:
Concluding Remarks
71. The issues discussed in this report seem to highlight failures in the regulatory process involving novel vaccines.
72. There is evidence of intentional manipulation of data.
73. Attempts were made to hide pertinent data from the public, even following a court order to release it.
74. These acts were even used to facilitate outright lying when rendering scientific conclusions.
75. Such acts compromise regulatory decision-making and rob the public of informed consent.
76. It should not be the responsibility of independent scientists to spend many months finding and reviewing complex, difficult-to-obtain, redacted documentation from around the world to identify these issues and then attempt to perform the critical functions of health regulators with substantial inefficiency.
77. So, a relevant question is, ‘how many other data and conclusions have been similarly compromised but remain undiscovered’?
78. The findings of this report raise serious questions about the integrity of the health regulatory process during the declared COVID-19 pandemic.
Report #2- Report on the Provenance of the Japanese Common Technical Regulatory Document
Request: I was asked by Dr. Robert W. Malone to provide a copy of the Japanese common technical document that I relied upon when making public statements and to comment on its provenance.
A copy of the common regulatory document that Pfizer/BioNTech provided to the Japanese government, and upon which Dr. Bridle relied when formulating his expert opinion, can be found at this link, marked Exhibit B. Specifically, this report had been submitted to Japan’s Pharmaceuticals and Medical Devices Agency.
In early 2021, Dr. Bridle had been meeting on an approximately weekly basis with several international groups of physicians and scientists with expertise relevant to COVID-19. One of these groups included Dr. Peter Doshi from the University of Maryland, USA. It was because of this scientific engagement that Dr. Bridle first became aware of the existence of the Japanese common regulatory document. Specifically, Dr. Doshi cited this document as reference #25 in an article that he published in the scientific journal The BMJ on May 18, 2021.
As an expert in preclinical vaccine studies, Dr. Bridle noted multiple, major concerns with the data set upon review. It was following discussion of these concerns with Dr. Doshi and several other scientists and physicians within the groups that Dr. Bridle was consulting with that he ultimately felt comfortable enough to publicly disseminate his findings. The consultation that occurred prior to public disclosure served as an informal peer review process.
Here is a screenshot taken from the paper published by Dr. Doshi showing the statement pointing to the Japanese common technical document (yellow highlighting added to the relevant text):

6. Here is a screenshot taken from the published paper showing reference #25:

7. Dr. Doshi’s paper was published on May 18, 2021. Here is a screenshot of the referenced webpage that was archived by the Wayback Machine4 on May 25, 2021, confirming that it was publicly available at the time of publication (note that the date can be found in the top, right corner of the image):
7. As of August 12, 2025, this link is no longer active (yellow highlighting added to relevant text):
8. This screenshot shows the page was still active as of March 26, 2022
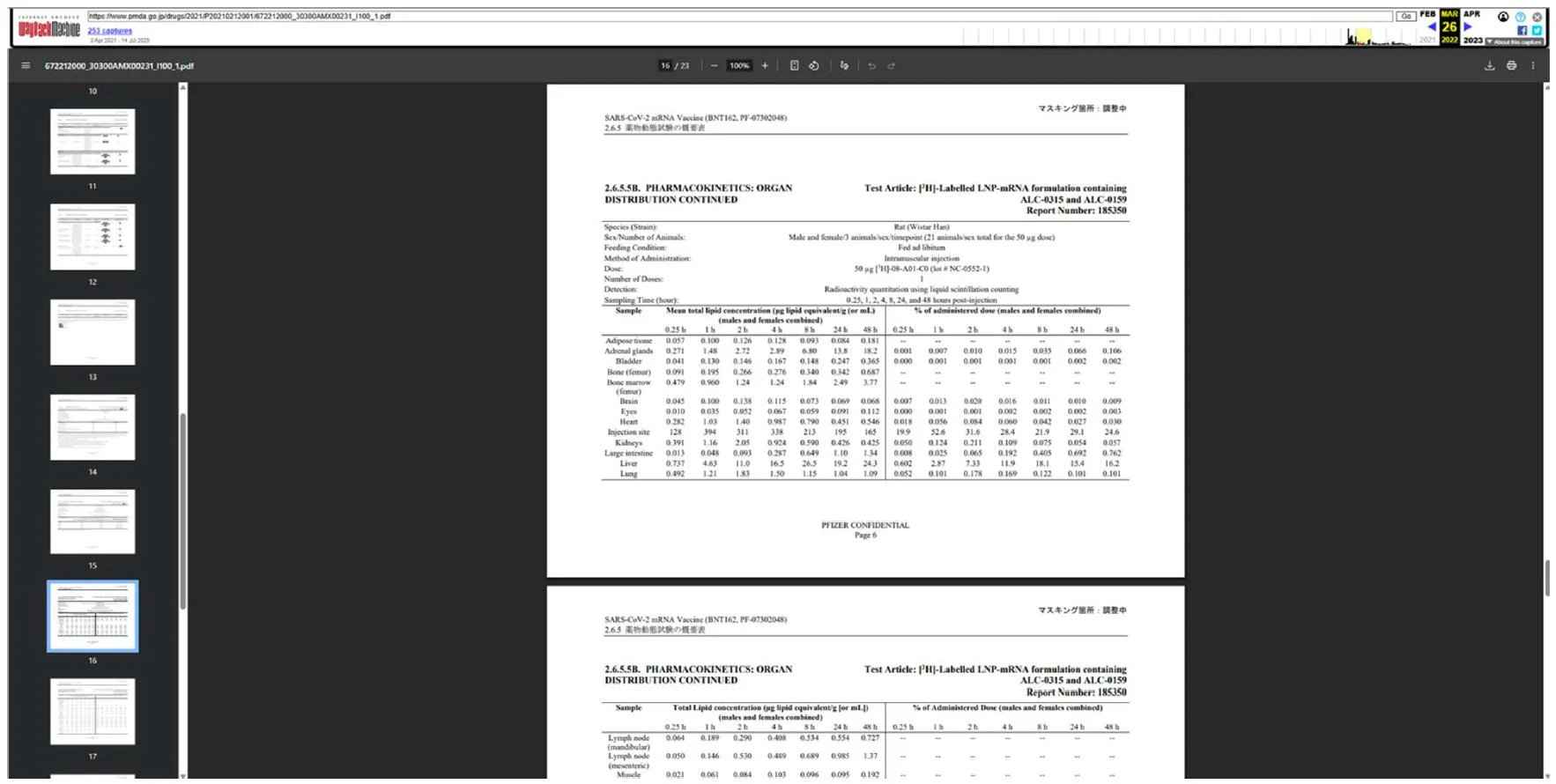
9. This screenshot shows the page was rendered inactive as of April 14, 2022

10. This screenshot shows there were no images archived between March 26, 2022, and April 14, 2022
