
What is Adulteration of pseudo-mRNA vaccines, and why should you care?
The FDA's job is to insure that drugs, vaccines, medical devices and foods are not adulterated. The remedy for adulteration is immediate recall and seizure if necessary.

Lately there has been a lot of discussion among insiders and those closely following the COVID “mRNA vaccine” story concerning contamination of the mRNA vaccines with DNA fragments which include DNA sequences derived from Simian Virus 40 (SV40).

What does this really mean? And why should you care?
Is this just another inside-baseball tempest in a teapot, akin to the various “social media expert/theorist”-promoted fringe conspiracy, fearporn-amplified controversies concerning graphene oxide , living hydras or snake venom in the vaccines, or that the lipid-pseudoRNA nanoparticles are actually 24th century Star-Trek science fiction nanobots which will reprogram all of our brains?
Is this DNA contamination/adulteration issue the real thing, one that should actually concern you- and the courts?
Drs. David Speicher, Kevin McKernan and colleagues are actually real life, bona fide serious scientific and technical experts in real-world application of sequence and molecular biologic analysis methodology. It is what they do, day in and day out, for a living. Which happens to be the specific technical area which they are reporting on.
These are not fringe “fever swamp” conspiracy theorists (Steve Bannon’s term).
Dr. David J. Speicher, University of Guelph Department of Pathobiology, 50 Stone Rd E, Guelph, ON, N1G 2W1, speicher@uoguelph.ca , ORCID 0000-0002-1745-3263
What Speicher et al are observing and reporting in this scientific manuscript linked below clearly demonstrates a profound failure of FDA and global regulatory authorities to do their most important job- to insure the purity and lack of adulteration of the pharmaceuticals which they authorize for marketing and use by physicians and allied health professionals.
At a minimum, it once again demonstrates the rampant willful blindness which seems to have pervaded the FDA/CBER vaccines branch under the “true believer” guidance of Dr. Peter Marks, who is neither a vaccine expert, nor an immunologist, nor a molecular biologist, nor someone who has any understanding of non-viral lipid nanoparticle-based polynucleotide delivery but rather is a clinical hematologist/oncologist that is the initial originator and continued proponent of the “operation warp speed” approach to vaccine (and now cancer drug) development. Which is to say bypassing almost all of the normal procedures and lessons learned from decades of development, manufacturing, approval for marketing and post-marketing surveillance of biological and drug products.
At worse, with this new information there is the appearance of a “smoking gun” demonstrating corrupt collusion between the US and other western administrative states’ pharmaceutical regulatory authorities and the pharmaceutical industry.
Based on my personal assessment of these data, this contamination appears to meet the formal criteria for pharmaceutical “adulteration”, which is strictly prohibited by US federal law. The prevention of drug, device and food “adulteration” is one of the central missions of the FDA - basically, a central reason that the FDA was created in the first place.
One key question which remains unresolved is how did this even happen?
Was this adulteration known by FDA, EMA, Paul Ehrlich Institute, Health Canada etc. and hidden from the public? If not known, how did this adulteration escape detection by virtually all western nation-authorized government regulatory experts?
Below is a screenshot of the tweet with a link to the associated pre-print manuscript which has launched this latest firestorm.

Abstract
Background: In vitro transcription (IVT) reactions used to generate nucleoside modified RNA (modRNA) for SARS-CoV-2 vaccines currently rely on an RNA polymerase transcribing from a DNA template. Production of modRNA used in the original Pfizer randomized clinical trial (RCT) utilized a PCR-generated DNA template (Process 1). To generate billions of vaccine doses, this DNA was cloned into a bacterial plasmid vector for amplification in Escherichia coli before linearization (Process 2), expanding the size and complexity of potential residual DNA and introducing sequences not present in the Process 1 template. It appears that Moderna used a similar plasmid-based process for both clinical trial and post-trial use vaccines. Recently, DNA sequencing studies have revealed this plasmid DNA at significant levels in both Pfizer-BioNTech and Moderna modRNA vaccines. These studies surveyed a limited number of lots and questions remain regarding the variance in residual DNA observed internationally.
Methods: Using previously published primer and probe sequences, quantitative polymerase chain reaction (qPCR) and Qubit® fluorometry was performed on an additional 27 mRNA vials obtained in Canada and drawn from 12 unique lots (5 lots of Moderna child/adult monovalent, 1 lot of Moderna adult bivalent BA.4/5, 1 lot of Moderna child/adult bivalent BA.1, 1 lot of Moderna XBB.1.5 monovalent, 3 lots of Pfizer adult monovalent, and 1 lot of Pfizer adult bivalent BA.4/5). The Vaccine Adverse Events Reporting System (VAERS) database was queried for the number and categorization of adverse events (AEs) reported for each of the lots tested. The content of one previously studied vial of Pfizer COVID-19 vaccine was examined by Oxford Nanopore sequencing to determine the size distribution of DNA fragments. This sample was also used to determine if the residual DNA is packaged in the lipid nanoparticles (LNPs) and thus resistant to DNaseI or if the DNA resides outside of the LNP and is DNaseI labile.
Results: Quantification cycle (Cq) values (1:10 dilution) for the plasmid origin of replication (ori) and spike sequences ranged from 18.44 - 24.87 and 18.03 - 23.83 and for Pfizer, and 22.52 – 24.53 and 25.24 – 30.10 for Moderna, respectively. These values correspond to 0.28 – 4.27 ng/dose and 0.22 - 2.43 ng/dose (Pfizer), and 0.01 -0.34 ng/dose and 0.25 – 0.78 ng/dose (Moderna), for ori and spike respectively measured by qPCR, and 1,896 – 3,720 ng/dose and 3,270 – 5,100 ng/dose measured by Qubit® fluorometry for Pfizer and Moderna, respectfully. The SV40 promoter-enhancer-ori was only detected in Pfizer vials with Cq scores ranging from 16.64 – 22.59. In an exploratory analysis, we found preliminary evidence of a dose response relationship of the amount of DNA per dose and the frequency of serious adverse events (SAEs). This relationship was different for the Pfizer and Moderna products. Size distribution analysis found mean and maximum DNA fragment lengths of 214 base pairs (bp) and 3.5 kb, respectively. The plasmid DNA is likely inside the LNPs and is protected from nucleases.
Conclusion: These data demonstrate the presence of billions to hundreds of billions of DNA molecules per dose in these vaccines. Using fluorometry, all vaccines exceed the guidelines for residual DNA set by FDA and WHO of 10 ng/dose by 188 – 509-fold. However, qPCR residual DNA content in all vaccines were below these guidelines emphasizing the importance of methodological clarity and consistency when interpreting quantitative guidelines. The preliminary evidence of a dose-response effect of residual DNA measured with qPCR and SAEs warrant confirmation and further investigation. Our findings extend existing concerns about vaccine safety and call into question the relevance of guidelines conceived before the introduction of efficient transfection using LNPs. With several obvious limitations, we urge that our work is replicated under forensic conditions and that guidelines be revised to account for highly efficient DNA transfection and cumulative dosing.
You can review the full manuscript yourself by following this link.
Understanding the Science behind this finding.
To follow the technical aspects and meaning of what has been discovered and demonstrated, you need to understand some molecular biology basics. I will do my best to explain and provide necessary context to those who have not had upper division molecular biology university training. I admit to being a little too close to the subject, and sometimes I assume too much background knowledge. If so, my bad. As Professor Richard Feynman is credited with saying, “If you can't explain something in simple terms, you don't understand it”. I will try to live up to his standards.
We have to start with the “central dogma” of biology. DNA makes RNA, RNA makes protein. <Yes, of course I know about reverse transcriptase, I was originally a retrovirologist, but lets table that for now please?>.
If you want to manufacture large amounts of pure RNA, you basically need to start with large amounts of DNA, and use a protein enzyme (bacteriophage T7 RNA polymerase in my original method, which is still used) plus RNA chemical subunits and a source of energy (ATP) to make RNA from the DNA. Then you need to break down the DNA into small fragments while still leaving the larger RNA intact. Then you need to purify the small DNA fragments from the larger RNA. In my original process, this was done using a type of filter (gel chromatography) which lets the little degraded DNA fragments and the small unused chemical subunits pass through more rapidly than the large RNA molecules. And then you throw away what comes out first- the little stuff (DNA fragments and unused chemicals) and keep the big stuff that comes through later - which is basically pure RNA dissolved in water.
Does that make sense?
Then, once you have that negatively charged purified RNA in water, you can make it more or less concentrated, mix it in fancy ways with other stuff like self-assembling positively charged fats to produce lipid nanoparticles, store it in a glass vial, and inject it into people. And that is the manufacturing process for pseudo-mRNA vaccines in a nutshell.
What could possibly go wrong, you ask?
In this case, at least two things appear to have gone wrong. The first involves the DNA that is used to manufacture the RNA <remember the central dogma of biology?>. And the second involves the DNA degradation and purification process employed <also as discussed above>.
There are apparently two different ways that were used to manufacture the DNA. The original manufacturing process used for the initial clinical trials employed the polymerase chain reaction, which can be and was used to make larger linear fragments of DNA (accuracy is somewhat problematic), which then were used to produce the RNA. This turned out to be too difficult, expensive, time consuming etc. to support mass manufacturing at the level needed to support worldwide dosing. So apparently Pfizer/BioNTech and Moderna both turned back to the original method which I used, which relied on circular “plasmid” DNA produced using bacteria (special lab strains of E. coli, which bacterium is commonly found in your gut).

You can think of plasmids as sort of like the purest forms of a bacterial virus. There are other more virus-like things that infect bacteria (called bacteriophage), but plasmids are circular DNA which can literally infect bacteria as pure DNA, and can direct those bacteria to transfer themselves and other plasmids from one bacteria to another.

These plasmids are like small parasitic DNA circles which often can help the bacterial host survive better under certain conditions such as exposure to antibiotics, and under those selection pressures the plasmids are maintained by the bacteria because they provide a survival or reproduction advantage. If the plasmid does not provide an advantage, other similar bacteria will outcompete those with the plasmid, because there is a cost to the bacterial host to maintain the parasite plasmid. <Be patient, this is going somewhere which is relevant to the main topic…>.
If you want to grow and recover (ergo manufacture) the most plasmid DNA that you can in a culture of E. coli bacteria, you want to use the smallest, most stripped down plasmid that can be engineered. Because any extra DNA sequences in the plasmid will come at a price of less plasmid production per liter in the resulting bacterial culture. Therefore you do not want to add DNA sequences into that plasmid that you don’t need for plasmid replication, antibiotic selection (Kanamycin or Neomycin in this case), and eventual RNA manufacturing. Does that part make sense to you?
So why, in heavens name, would any corporation developing and deploying a plasmid-based manufacturing process for large scale synthesis of RNA from a DNA template include sequences in the plasmid that are not necessary for the intended purpose? Why add sequences lifted from a known oncogenic (ergo, cancer causing) DNA virus like Simian Virus 40 (SV40)?
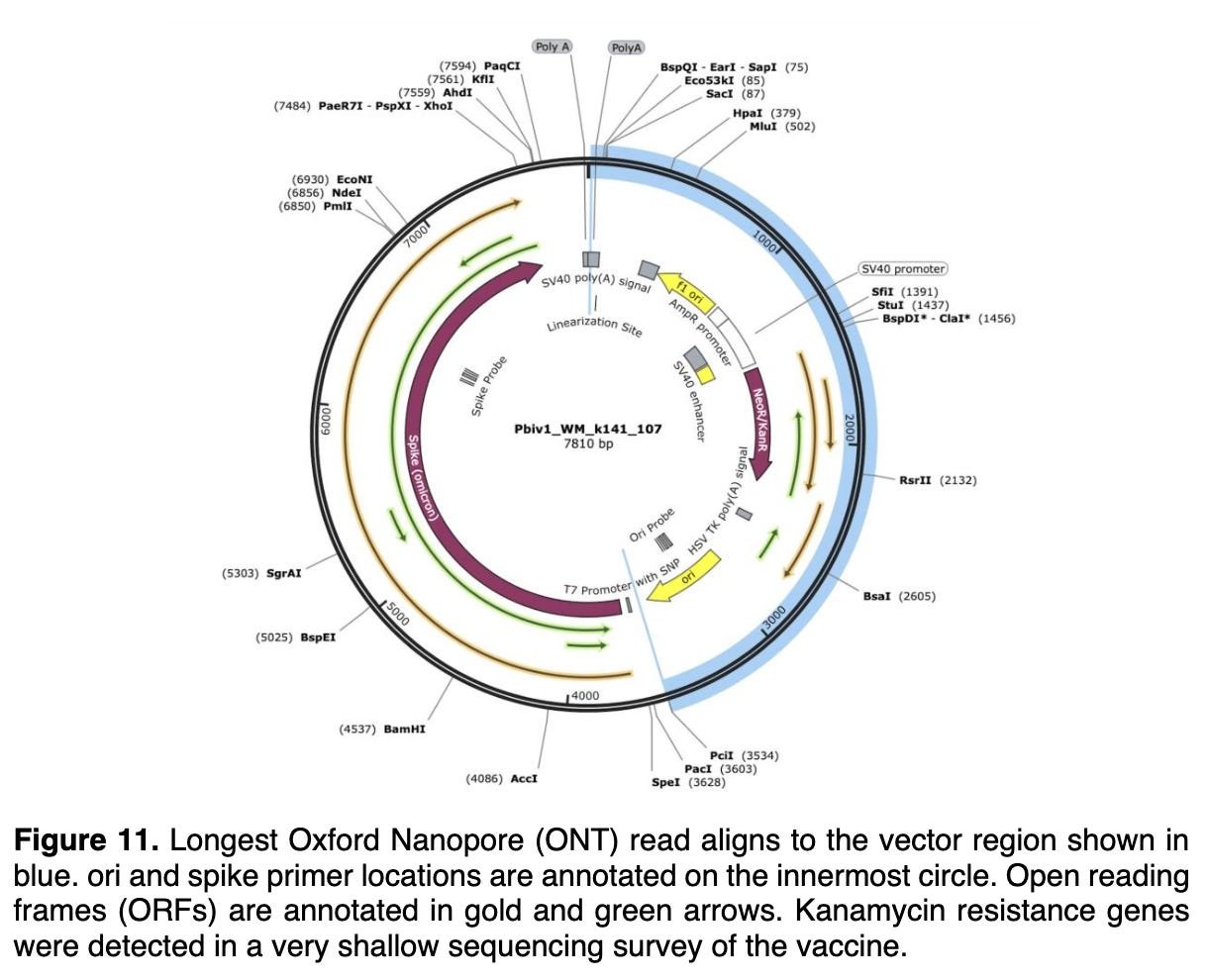
It turns out that these specific SV40 sequences which have been identified in the plasmid DNA fragment contamination documented (above) by Speicher et al are commonly used in a specific type of engineered bacterial plasmid which was developed decades ago for use by molecular biologists. This is well established “common core” recombinant DNA technology.
Bacterial plasmids can and have long been engineered to replicate and produce RNA (and proteins) in both bacteria and in animal cells. Such plasmids are called “shuttle vectors” in the industry. They can be manufactured and purified in large quantity using laboratory E. coli strains, and then transferred (“transfected”) into animal cells where they can replicate for a period of time (under some conditions) and produce the RNA and protein of interest in the animal cells- under the control of promiscuous SV-40 derived sequences in this case.
So what the heck are the SV-40 sequences doing in plasmids whose sole purpose is to be purified and used to produce large amounts of RNA “in the test tube” using a commercial-grade enzyme-based manufacturing process? Good question.
I can speculate or hypothesize, but I suggest that it is the job of Mr. Pharma and Mr. Government Regulator to answer that question. And to address why this was never disclosed to the public, let alone subjected to any formal assessment of possible risks when small fragments of these SV-40 and other plasmid DNA sequences (including antibiotic resistance gene fragments) are delivered into the bodies of patients using the most efficient systemic in-vivo non-viral delivery technology ever developed in the history of the world.
Can I imagine possible risks?
In short, yes. One way or another, at a minimum such fragments are likely to impact on gene expression in the human cells that take up the DNA. One possible impact could involve development of cancers - what molecular biologists and cancer researchers would call transformation (note the emphasis). Should these risks have been investigated before any of this was allowed to proceed and be injected into human beings (without their knowledge)? Of course they should have. And also self evident is this should all have been disclosed to all concerned. If the FDA, EMA, Paul Ehrlich Institute, Health Canada etc. were not informed, then this would be fraud. If they were informed and did nothing, than that would be criminal negligence <in my opinion, but I am an MD, not a JD>.
I predict a hurricane of factchecker propaganda, obfuscation and whaddaboutism raised about this, but the core facts are indisputable.
So, there is that.
Rimshot.

Professor Feynman, I hope I have met or exceeded your expectations.
Then what’s Adulteration got to do with it?
(apologies to Tina Turner)
“Adulteration” is a legal regulatory term, widely used in the US Code of Federal Regulations (CFR) which provide the congressional authorization and mandated scope for the US Food and Drug Agency. See for example 21 U.S. Code § 351 - Adulterated drugs and devices.
According to Websters:
The meaning of ADULTERATE is to corrupt, debase, or make impure by the addition of a foreign or inferior substance or element; especially : to prepare for sale by replacing more valuable with less valuable or inert ingredients.
According to the US Congress, Adulteration is defined (in CFR Title 21, CHAPTER 9, SUBCHAPTER V § 351) as follows (partially redacted for focus and simplification):
A drug or device shall be deemed to be adulterated—
(a)Poisonous, insanitary, etc., ingredients; adequate controls in manufacture
(B) if it is a drug and the methods used in, or the facilities or controls used for, its manufacture, processing, packing, or holding do not conform to or are not operated or administered in conformity with current good manufacturing practice to assure that such drug meets the requirements of this chapter as to safety and has the identity and strength, and meets the quality and purity characteristics, which it purports or is represented to possess.
(b)Strength, quality, or purity differing from official compendium
If it purports to be or is represented as a drug the name of which is recognized in an official compendium, and its strength differs from, or its quality or purity falls below, the standard set forth in such compendium. Such determination as to strength, quality, or purity shall be made in accordance with the tests or methods of assay set forth in such compendium, except that whenever tests or methods of assay have not been prescribed in such compendium, or such tests or methods of assay as are prescribed are, in the judgment of the Secretary, insufficient for the making of such determination, the Secretary shall bring such fact to the attention of the appropriate body charged with the revision of such compendium, and if such body fails within a reasonable time to prescribe tests or methods of assay which, in the judgment of the Secretary, are sufficient for purposes of this paragraph, then the Secretary shall promulgate regulations prescribing appropriate tests or methods of assay in accordance with which such determination as to strength, quality, or purity shall be made.
(c)Misrepresentation of strength, etc., where drug is unrecognized in compendium
If it is not subject to the provisions of paragraph (b) of this section and its strength differs from, or its purity or quality falls below, that which it purports or is represented to possess.
(d)Mixture with or substitution of another substance
If it is a drug and any substance has been (1) mixed or packed therewith so as to reduce its quality or strength or (2) substituted wholly or in part therefor.
What is the legal remedy for Adulteration?
See the FDA document CPG Sec. 420.100 Adulteration of Drugs Under Section 501(b) and 501(c) of the Act. *Direct Reference Seizure Authority for Adulterated Drugs Under Section 501(b)* .
Section 501(b) of the Food, Drug, and Cosmetic Act (the Act) deems an official drug (i.e., a drug purported to be or represented as a drug the name of which is recognized in an official compendium) to be adulterated if it fails to conform to compendial standards of quality, strength or purity. Compendial tests or assay methods are used when determining such conformance under 501(b); the standards are stated in individual monographs as well as portions of the General Notices section of the USP/NF. Standards and test methods have been established for such characteristics as potency, sterility, *dissolution*, weight variation and content uniformity. If an official drug fails to conform to one or more compendial standards of strength, quality or purity, but plainly states on the label how it differs from the standard, then the drug is not deemed to be adulterated under Section 501(b).
POLICY: Any official drug which, when tested by compendial methods, fails to conform to compendial standards for quality, strength, or purity, is adulterated unless the differences from such standards are plainly stated on the drug's label. Any *drug which is not recognized in an official compendium is adulterated if its strength differs from, or its purity or quality falls below that which it purports or is represented to possess, when tested by scientifically sound methods.*
REGULATORY ACTION GUIDANCE: Recommendations for regulatory action will be considered in the above instances of adulteration. The regulatory action of choice will depend upon the circumstances of each case.
In cases where there is a health hazard, the first choice of action should be recall, particularly for drugs found to be non-sterile, and for narrow therapeutic range drugs that fail potency or dissolution tests. However, where the appropriate division within the Office of Pharmaceutical Quality Operations (OPQO) has advised the firm of such a defective product, and the firm fails to recall, seizure should be considered.
Seizure recommendations charging adulteration under section 501(c) should be submitted to the Office of Compliance, Center for Drug Evaluation and Research (HFD-300) (CDER).
Appropriate division offices within OPQO are authorized to submit seizure recommendations, charging adulteration under section 501(b), directly to the OPQO Program Director and Office of Enforcement without CDER review under the following circumstances, provided introduction or delivery for introduction into interstate commerce has been documented:
1. An official sample of either a compendial bulk pharmaceutical chemical or a compendial finished dosage form has been analyzed, using the compendial methods without modification and found to fail both the original and check analyses.
2. The analyzing laboratory has certified in the transmittal memorandum that an unmodified compendial method was used. Note: No tolerance need be applied beyond that provided by the official compendium. 3. For sterile products, no check analysis is needed provided the compendial sterility test was utilized without modification, the product is one that is required to be sterile, and all relevant laboratory controls (including positive and negative) are satisfactory. Where the analyzing laboratory deviates from the official compendial analytical method(s), a detailed description of the deviation(s) and justification for such deviation(s) can be submitted to CDER for review. In such cases, CDER will review only the deviation(s) and not the choice of regulatory action or other documentation. For seizure actions, the charges may be drafted as follows: That the article of drug was adulterated, when introduced into and while in interstate commerce, and is adulterated while held for sale after shipment in interstate commerce, within the meaning of 21 U.S.C. 351(b), in that it purports to be and is represented as a drug, the name of which is recognized in an official compendium (United States Pharmacopeia) and its strength differs from, and its quality and purity falls below the standard set forth in such compendium because it fails the official (INSERT TYPE OF TEST) test.
or
That the article of drug was adulterated, when introduced into and while in interstate commerce, and is adulterated while held for sale after shipment in interstate commerce within the meaning of 21 U.S.C. 351(c), in that it is a drug not subject to the provisions of 21 U.S.C. 351(b) and its strength differs from, and its purity and quality falls below, that which it purports or is represented to possess because (e.g., the drug contains less than the amount of (INSERT NAME OF INGREDIENT) on the label). It should be kept in mind that the types of adulteration found under 501(b) and 501(c) may be indicative of a wider problem involving failure of the manufacturer to adhere to current good manufacturing practice that should be addressed.
First, as discussed above, it is my expert professional opinion that, to the best of my knowledge (assuming the data and facts reviewed above to be true) that the products tested by Speicher et al meet the formal regulatory criteria for adulteration.
Secondly, it is my expert professional opinion that, to the best of my knowledge (assuming the data and facts reviewed above to be true) that the products tested by Speicher et al represent a significant risk of health hazard. Furthermore the safety data accumulated by CDC VAERS, VSAFE, Pfizer Pharmacovigilance, and safety databases from throughout the world demonstrate that there is reasonable evidence that these products represent a significant risk of health hazard, and that based on both CFR Title 21, CHAPTER 9, SUBCHAPTER V § 351 and FDA guidance CPG Sec. 420.100 Adulteration of Drugs Under Section 501(b) and 501(c) of the Act. *Direct Reference Seizure Authority for Adulterated Drugs Under Section 501(b)* , these products should be subject to immediate recall.
Third and finally, in the event that these products are not recalled by the sponsor (noting that, in the special case of EUA, the sponsor is the FDA) it is my expert opinion that seizure should be strongly considered by the States Attorney General for each of the 50 of the United States.
Signed this Monday 23 October, 2023

If you would like these blog posts to appear in your email stream,
please sign up to